
Phäochromozytom
Phäochromozytome stammen von den chromaffinen Zellen im Nebennierenmark ab. Sie werden aufgrund ihrer Beziehung zu den Paraganglien als Paragangliome bezeichnet. Extraadrenale Paragangliome oder Phäochromozytome lassen sich aber prinzipiell überall dort finden, wo sympathisches Nervengewebe vorhanden ist.
Epidemiologie
Die Inzidenz beträgt ca. 2 pro 1 Mio. Einwohner und Jahr. Diese Angaben sind allerdings wahrscheinlich zu niedrig, da Phäochromozytome häufig der Diagnostik entgehen. Autopsiestudien zeigen eine 5mal höhere Inzidenz von dieses Tumors.
Assoziierte endokrine Erkrankungen
- MEN I-Syndrom
- MEN II-Syndrom (C-Zell-Karzinom, Nebenschilddrüsenhyperplasie)
- von Hippel-Lindau-Erkrankung
- Neurofibromatose
- Carney-Trias (Leiomyosarkom des Magens, Lungenchondrom und extraadrenales Phäochromozytom)
Pathophysiologie
Der Vorläufer für die biogenen Amine ist die Aminosäure Tyrosin, die durch das Enzym Tyrosinhydroxylase zu Dopa transformiert wird. Aus Dopa wird durch Transformation Dopamin und Noradrenalin gebildet, welches in intrazellulären Granula gespeichert ist. Nur Tumoren des Nebennierenmarks können Adrenalin zusammen mit Noradrenalin produzieren, da das bestimmende Enzym cortisolabhängig ist. Extraadrenale Tumoren stellen daher nur Noradrenalin und/oder Dopamin her.
Symptome
Zirka die Hälfte der Patienten mit Phäochromozytomen haben typische Hochdruckattacken, die oft durch physische Anstrengungen verursacht werden. Der Blutdruck kann dabei Werte über 300 mmHg annehmen. Die Attacken dauern zwischen 5 und 10 min. Sie sind assoziiert mit Kopfschmerzen, Herzrasen, auffälliger Blässe und Kaltschweißigkeit. Bei einem Teil der Patienten kommt es nicht zu anfallsartigen Blutdruckkrisen, sondern es liegt eine Dauerhypertonie vor.
Diagnostik
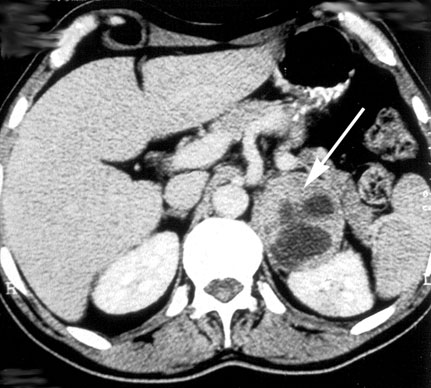
Typisch sind erhöhte Katecholaminkonzentrationen im Urin. Als sensitivster Parameter hat sich die Metanephrinausscheidung im 24h-Sammelurin erwiesen. Dennoch sollten auch Adrenalin, Noradrenalin und Normetanephrin bestimmt werden. Die diagnostische Aussagekraft wird ggf. durch die Wiederholung der Untersuchung erhöht. Die Serumkatecholamine sind sehr empfindliche Parameter, die leicht durch eine nicht korrekte Blutabnahmetechnik verfälscht werden können.
Als besonders geeignetes bildgebendes Verfahren ist die MIBG-Szintigraphie zu nennen, die eine für das Phäochromozytom hochspezifische Untersuchung ist und ca. 80-90 % aller Phäochromozytome darstellt. Bei typischer Klinik und Laborkonstellation wird der Tumor jedoch zumeist mittels Schnittbilddiagnostik (CT oder MRT) dargestellt.
Präoperative Behandlung
Ist die Diagnose biochemisch gestellt, sollte vor einer operativen Behandlung eine Vorbehandlung mit Alphablockern erfolgen. Hierzu wird gewöhnlich Phenoxybenzamin eingesetzt. Das Medikament wird in aufsteigender Dosierung appliziert, bis es zu Nebenwirkungen (orthostatische Dysregulation, Gefühl der verstopften Nase) kommt. Während dieser Phase sollte der Patient viel trinken, gelegentlich sind auch Infusionen erforderlich, um den erheblichen Volumenmangel dieser Patienten auszugleichen.
Operation
Phäochromozytome können konventionell oder endoskopisch entfernt werden. Bis zu einer Größe von ca. 6 –8 cm ist der endoskopische Zugang das Verfahren der Wahl.
Malignes Phäochromozytom
Diese Diagnose kann nur aufgrund einer lokalen Tumorinvasion oder das Nachweis von Metastasen gestellt werden. Etwa 5-10 % der Phäochroozytome sind maligne. Der klinische Verlauf der Krankheit kann sowohl schnell und aggressiv als auch langsam und eher gutartig sein. Die 5-Jahres-Überlebensrate liegt zwischen 35 und 60%.
Symptomatische Therapie
Die Therapie sollte chirurgisch sein. Bei nichtresektablen symptomatischen Tumoren oder Tumormetastasen ist eine medikamentöse Langzeittherapie mit Alpha-Blockern indiziert. Über die Behandlung mit Chemotherapeutika gibt es bisher nur wenig Daten.